

Photo credit to Gerd Altmann from Pixabay
This is more of a collection of notes than an article. I was trying to understand a bit more about ASCVD and fell down a rabbit hole for a month.
Arteriosclerosis translates to "hardening of arteries". The loss of elasticity is a key component of pathogenesis that links these diseases. Usually, this is considered a family of 3 types: One, arteriolosclerosis, is a hardening of small arteries/arterioles; causes downstream ischemia, rarely other problems. Two, Monckeberg medial sclerosis, is defined by calcification of the walls of muscular arteries; calcification does NOT intrude on the lumen and is usually of no clinical significance. Three, atherosclerosis, the most common, the most important, and the subject of the remainder of this article [1].
Atherosclerosis is the primary pathogenic event leading to negative cardiovascular events (stroke, heart attack, etc). As such, it is the primary pathology underlying approximately half of all deaths in the Western world [1]. In short, a plague of lipid, cholesterol, clotting agents, immune system cells, and cell detritus forms on or just under the enthothelium of the artery. This occludes the artery leading to ischemic injury downstream. More disastrously, the portions of the plaque, or the thrombus which can form on top of the plaque, can dislodge from the wall into the blood stream. Once dislodged, it may trigger a clotting cascade, growing as it travels down into smaller arteries/arterioles. Eventually reaching a vessel too small to traverse, it becomes mechanically stuck, catastrophically blocking the artery supply to downstream tissues. In the brain this is an ischemic stroke, in the heart a myocardial infarction, and in the lung a pulmonary embolism. All of these may be fatal events, depending on the artery blocked, treatment provided, and resilience of the entire organism.
Most of the strategy around atherosclerosis involves slowing it down. This means treating the early stages, lowering the concentration of atherogenic species in the blood by lifestyle or pharmacology. I found much more information on the mechanisms of atherogenesis at the early stages (LDL, HDL, cholesterol esters, apolipoproteins, etc) than I did on the later stages (inflammation cascades and calcification). There seem to be fewer drug targets in the later stages of the disease. The main exception is coagulation cascade. By using blood thinners, doctors can reduce the thrombus formation and improve cardiovascular outcomes in patients with known mid-late stage atherosclerosis.
Gabriele Falloppio, or "Fallopius", was a famed 16th century Italian anatomist. He was supposedly the first to describe a transition of arterial wall to bone in 1575 (see confusion below). "Atheroma" was first used by Albrecht von Haller (Austria/Swiss pathologist) in 1755 to describe lacerated rotten arteries [2,3]. The role of inflammation in atheromatous lesions was recognized as early as 1858 [4] by Virchow, the Polish/German "founder of cellular pathology" [5]. Atherosclerosis was first named in 1904 [6,7]. In 1913, it was discovered that cholesterol played a decisive role in the development of atheromatous lesions [8,9], one of the great cardiology breakthroughs of the 20th century [10,11]. The "no atherosclerosis without cholesterol" thesis came from Anichkov's (Russian pathologist) experiments feeding rabbits a cholesterol rich diet [12]. By 1924, it was hypothesized that the early and late stage lesions were different points in the disease progression using a histological categorization which persisted until the late 20th century [13,14].
Many articles cite Fallopius's observation of arteries degenerating to bone as originating in the year 1575 [15,16,17,18]. Fallopius himself certainly did not write anything about arteries in 1575 because he lived from 1523 to 1562, dying 13 years before the claimed observations on "arteries to bone" [19]. Things can obviously be published posthumously, and in the case of Fallopius, that is what happened with all but one of his books. However, I am skeptical that most of the above authors saw even a translation of the original document because I cannot find any reference to the specific publication where this was mentioned and because it is frequently referred to an "observation in 1575" rather than "published in 1575." For now I assume the observation was in [20] based on the publication date but I have not laid eyes on a translation so I do not know for sure.
Atherosclerosis is typically found in medium to large arteries, mostly the coronary arteries, carotid arteries, ilio-femoral arteries, and aorta [21,22]. To understand how a plaque forms, we must first understand the structure of a blood vessel.
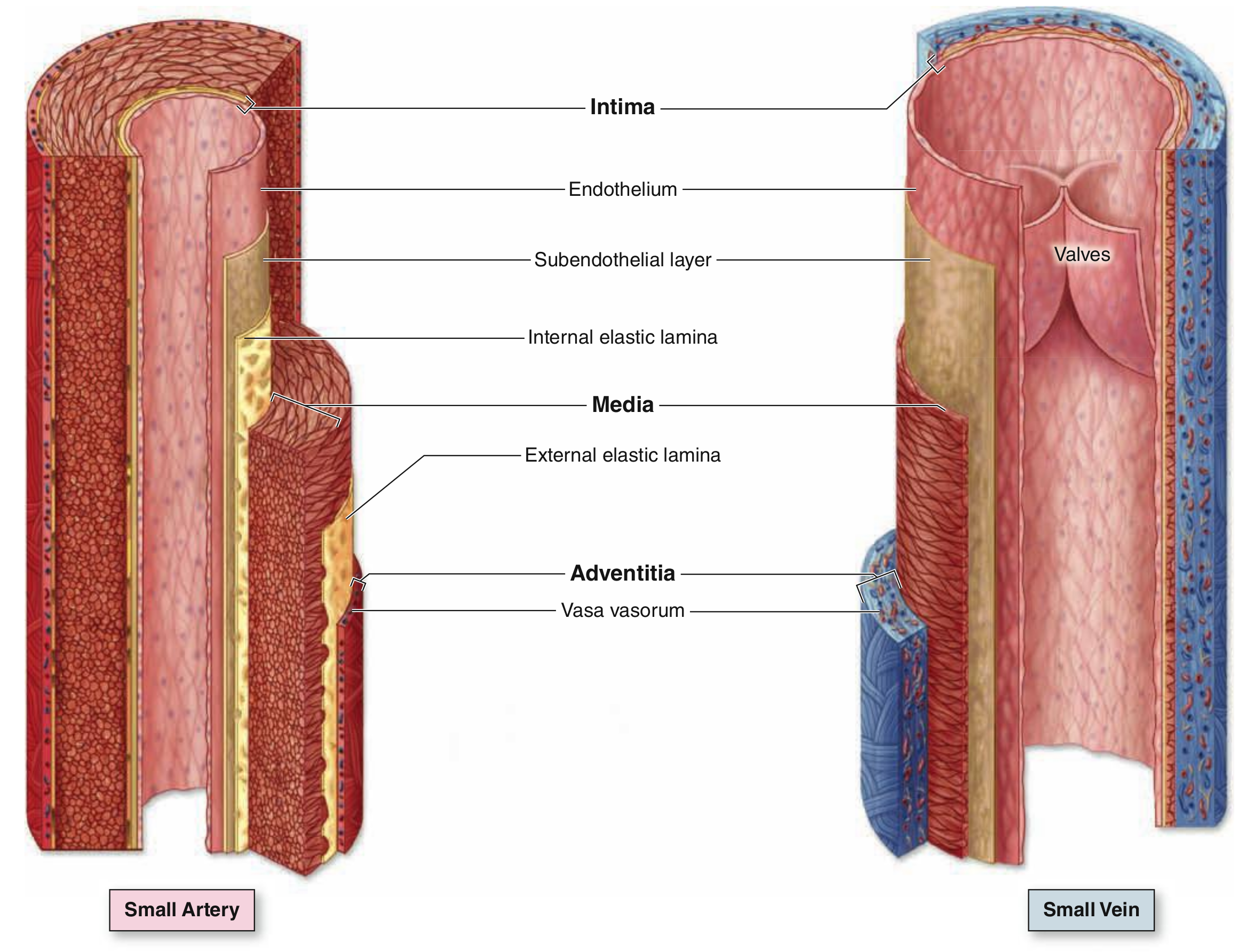
[Caption] Image from page 221, [23]. The exact dimensions vary with the size of vessels but a combined intima-media thickness for most arteries is rarely greater than 700 microns in the absence of pathology. The large vessels, like the aorta, have a combined average intima-media thickness of 6 +- 2 mm in healthy young adults [24]. Though most often measured together, the media is usually the majority of that thickness. In arteries, the adventitia is typically under 200 microns.

[Caption] Histology slide, cross section of artery wall, image from page 149, [25]. Intima, media, and adventitia are labeled I, M, and A, respectively. The vasa vasorum, labeled V, is the network of blood vessels which provide blood supply to the structure of the large vessels. Vasa vasorum translates roughly from Latin to "vessels of vessels".
Arteries have three layers: tunica intima, tunica media, and tunica adventitia, or just intima, media, and adventitia. The intima contains the endothelium, some smooth muscle fibers and an internal elastic lamina (composed of extracellular matrix of the protein elastin). As the name suggests, this gives elastic responses to mechanical deformation of the vessel. Moving away from the lumen, the media is a series of concentric layers of helical smooth muscle cells. The extracellular elastic fibers, reticular fibers and proteoglycans are secreted by the smooth muscle cells. Some arteries have an external elastic lamina separating the media from the adventitia. The adventitia layer is Type I collagen and elastic fibers. It is bound to the stroma of the adjacent organ. [23,25]
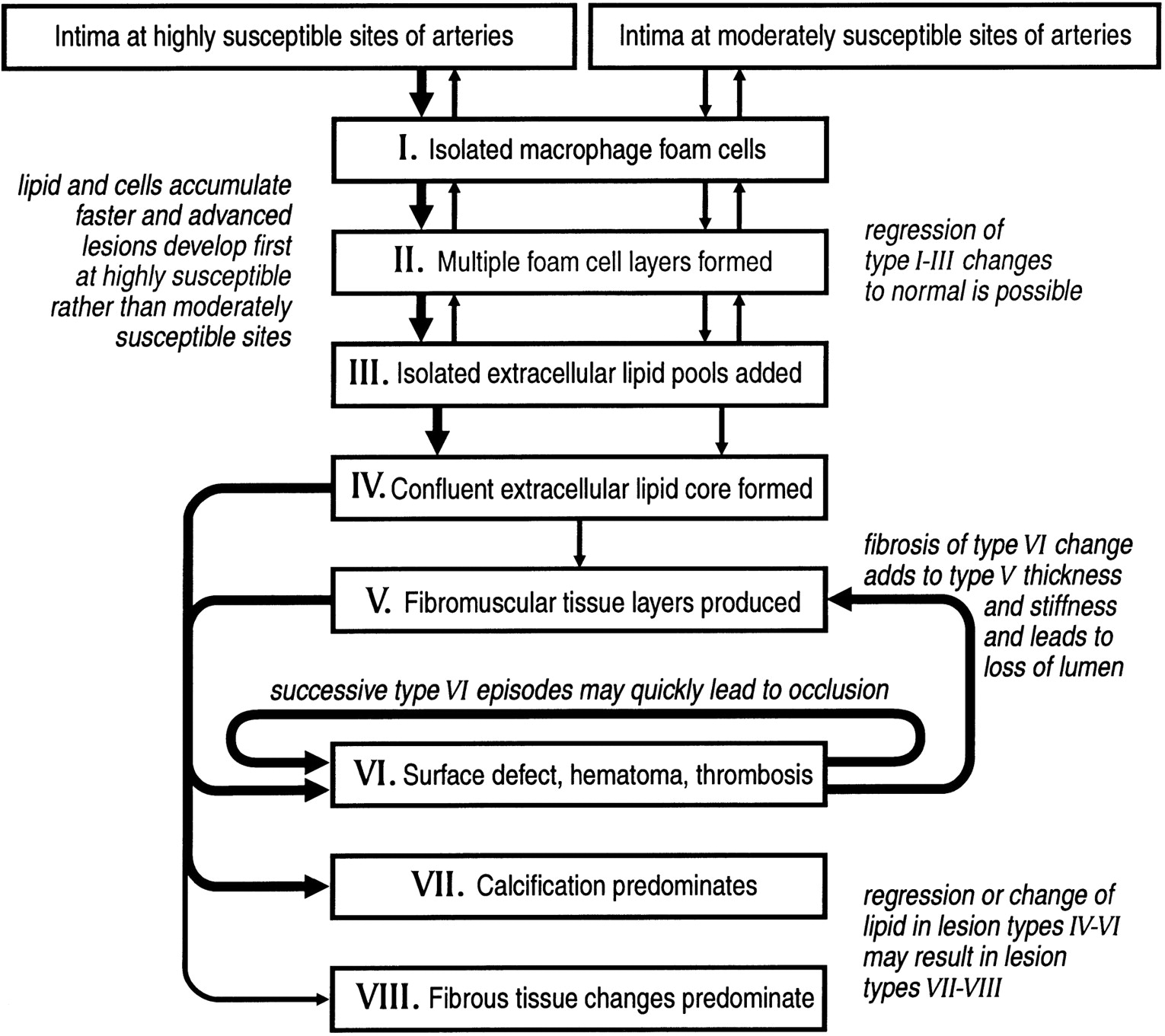
[Caption] Schematic of the progression and defining characteristics of atherosclerotic plaques. Copied from [26].
When discussing the composition of an atherosclerotic plaque, it is important to recognize that the composition is a function of disease state. As plaques progress, compositions change depending on the environment and environmental history during plaque development. One would not expect that a plaque grown in an artery primarily driven by hypertension would have no distinguishing features from a plaque driven by hyperlipidemia. Furthermore, size is not the only factor which differs between a plaque just beginning to grow in a 25 year old and one about to rupture in an 80 year old.
In the literature going back to at least the 1990s, plaques are identified as "early lesions", which are subdivided into Type I, Type II, and sometimes Type III. Whether a Type III lesion is "early" is the semantically debatable part. Whether we interpret "early" to mean advance stage of the disease (that a lesion starts "early" before progressing to "late" stage disease) or "early" meaning early in life, we are not exactly right. It is not conclusive that advanced lesions must have been preceded by early stage lesions matching these descriptions or that only the elderly experience "late" stage lesions. However, there is strong evidence to suggest most lesions develop slowly over decades from Type I to Type V and statistically speaking most of the later stage lesions are found in older populations.
"Type I lesions consist of the first microscopically and chemically detectable lipid deposits in the intima and the cell reactions associated with such deposits." - [27] Type I lesions are typically not visible to the naked eye. They are defined by the presence of small groups of macrophages full of lipid droplets (known as foam cells) in the intima of the artery. These are sometimes present in infants (45% of infants at 8 months have histologically detectable lesions [27]).
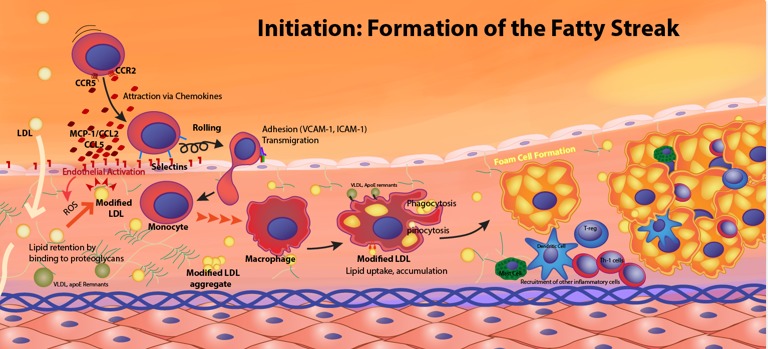
[Caption] Description of the development of a fatty streak from [28].
Type II lesions have had many names (roughly ordered by most modern first): type II [27], "fatty streak" [23,27], "sudanophilic deposits" or "sudanophilic lesions" [29], "sudanophilia" [30,31,32]. The sudan names originate from the Sudan III and Sudan IV dyes used to stain fatty deposits bright red in histology sections.
Type II lesions are often (but not necessarily) visible to the naked eye as yellow colored patches or spots when viewing the dissected artery from the luminal side. Lesions are Type II by definition when their dominant volume is macrophage foam cells in stratified layers. Intimal smooth muscle cells also contain histologically visible lipid droplets. Some mast cells and T-lymphocytes may also be present. There may be extracellular lipid droplets and vesicles visible with electron microscopy in addition to some lipoprotein particles. However, the vast majority of lipid is contained within the foam cells. [27]
There are a heterogeneous mix of lipids and cholesterol esters in atherosclerotic lesions. Using desorption electrospray ionization mass spectroscopy, studies have detected over 25 separate lipid species in a single plaque [33]. Over 150 separate lipids have been found in human atherosclerotic plaques; most of these are cholesterol esters, sphingomyelins, phosphatidylcholines, and lysophosphatidylcholines [34].
This lesion is defined as not quite Type II and not yet Type IV. It is sometimes called an "intermediate lesion", a "transitional lesion", or a "preatheroma". Histologically, a Type III lesion is defined by extracellular, microscopically visible lipid droplets interspersed around the smooth muscle of intima which may be membrane-bound or free. These lipid pools disrupt the structure of intimal smooth muscle cells. Compared to Type II, Type III lesions have more free cholesterol, fatty acid, sphingomyelin, lysolecithin, and triglyceride. At this stage, there is no macroscale separated lipid core in the lesion. [27]
Type IV are the first of the late stage atherosclerotic plaques and the lowest level designated as a "atheroma" [27] or "fibrous plaque" [14]. The Type IV lesion is primarily distinguished by the formation of an extracellular lipid core [14,26]. The lipid core forms as the extracellular droplets coalesce [35].
Despite thickening the arterial wall clearly to the unaided eye, Type IV lesions do no necessarily obstruct the lumen (i.e. they grow into the tissue not into the lumen) [36].
As the lesion develops into a Type V, it will grow a fibrous cap of mostly collagen. However, because the cap in Type IV and Type V is indistinguishable in standard histological sections, or with the unaided eye, the lesion may also be called a "fibrous plaque" at this stage. To be clear, there is no fibrous cap on a Type IV lesion (if there was, it would be a Type V) but because of visual similarities, the naming conventions overlap [14]. Type IV and V lesions are clinically relevant because they are prone to developing fissures, hematomas, and thrombi [14].
As the previous types of lesions go by many names, so too Type V may be called a "fibroatheroma", "fibrolipid plaque", or a "fibrous plaque" [14]. As of the early 2000s, the subtypes of Type V lesions, Type Va, Vb, and Vc defined in [14] have been redefined in [26] as Type V, VII, and VIII respectively. The a, b, and c subtypes are a depreciated nomenclature according to the original author [26].
Type V lesions are defined by the presence of a fibromuscular cap composed of mostly collagen (the "fibro" part) and endoplasmic reticulum-rich smooth muscle cells (the "muscular" part) which do not orientate themselves as they would in a healthy intima. Often this development is accompanied by a narrowing of the arterial lumen. Type IV and V lesions are clinically relevant because they are prone to developing fissures, hematomas, and thrombi [14].
The Type V categorization includes many subtype morphologies such as a series of lipid cores separated by thick layers of fibrous connective tissue ("multilayered fibroatheroma"). There are several competing theories on the mechanism of formation: (theory 1) formed by the mechanical forces which change as the plaque thickens, (theory 2) formed by the repeated thrombotic deposits followed by extracellular lipid and macrophage foam cells accumulating between the new fibrotic layer and the endothelium. I have not been able to find a link between formation mechanism and prognosis/treatment/clinical utility but I'm sure this is an active area of research [37].
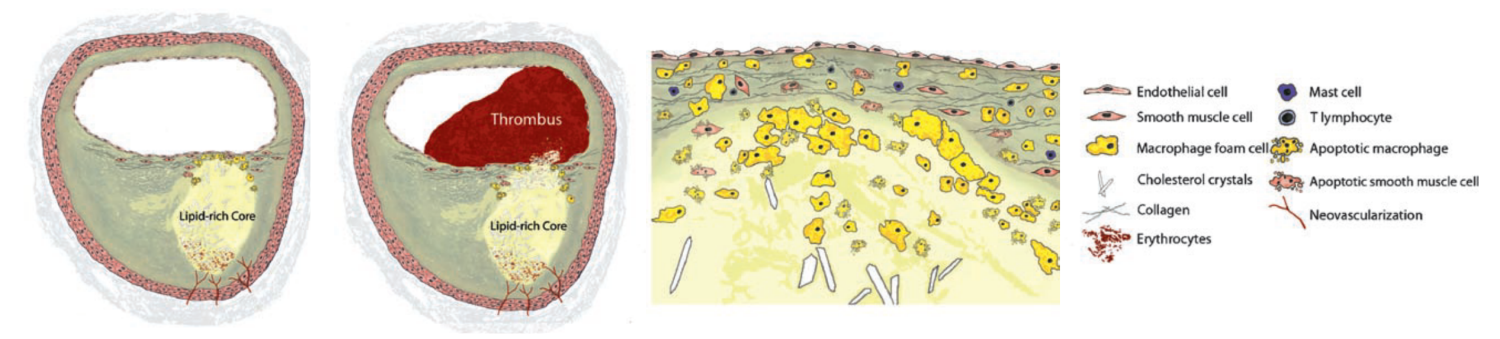
[Caption] Schematic from [38] showing a Type V lesion (far left) and Type VI lesion (center left). The defining feature of the Type VI is the thrombus in the arterial lumen. Notice the gradient of apoptotic cells along the radial direction from the center of the lipid core.
Aliases for Type VI lesion include "complicated lesion" and "complicated plaque" [14].
A Type VI lesion is defined by a surface defect which creates a hematoma or thrombus (blood clot outside or inside the circulatory system, respectively). It is not necessary for a lesion to move linearly through the Type classifications. A Type IV or Type V can transition straight to a Type VI or VII.
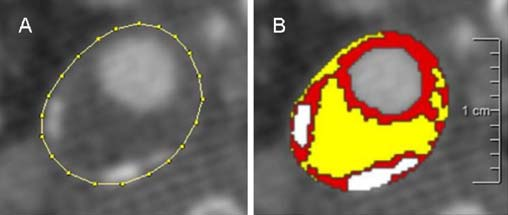
[Caption] (Image and caption directly copied from [39]) "Semi-automatic assessment of plaque component areas in MDCT (multidetector computed tomography) images. Axial MDCT image of an atherosclerotic carotid plaque; the region of interest is drawn on the outer vessel wall (a). Ranges of Hounsfield unit (HU) values represent three different plaque components: yellow lipid core (<60 HU), red fibrous tissue (60–130 HU), and white calcification (>130 HU) (b)." HU is the standard measure of radiographic absorption of the medium being imaged. See Wikipedia. Because of the predominance of calcium, this is a Type VII lesion.
Other names for a Type VII lesion: "Calcified plaque", "advanced lesion" [14]. Type VII lesions are defined by the presence of calcium crystals which may completely take over the extracellular lipid and remnants of dead cells. Like in bone, the calcium is trapped in hydroxyapatite, \(\require{mhchem} \ce{Ca5(PO4)3OH}\) [40], in a hexagonal structure [41].
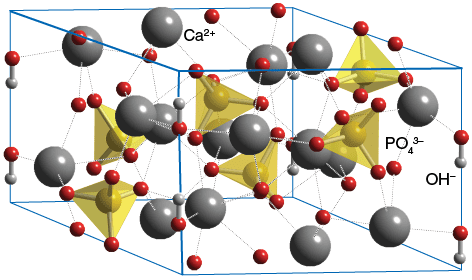
[Caption] Image from ChemTube3D. Hydroxyapatite crystal structure.
Coronary artery calcium (CAC) is a relative measure of arterial calcification based on the number of arterial voxels (volumetric pixels) in a CT scan above a threshold number [42]. The Agatston method of computing the score weights the radiographic density of the voxels in the calculation but is otherwise the same method [43]. Since these methods are not naturally bounded, they are used to compute risk stratification along with age, sex, race, and other lab metrics. Because the CAC score detects late stage lesions, a CAC score of 0 does not indicate 0 risk of ASCVD. The value of CAC scores is highly context dependent and sometimes counterintuitive. For example, high dose statins can increase CAC scores, possibly because the calcification process is a progression of healing that is enabled by the lower cholesterol burden [44].

[Caption] Coronary artery calcium scores in age and race population of by percentile. Plot generated from Table 2 in [45]. White men age 75-85 have a mean CAC score of 2900 and 4600 for the 90th and 95 percentile, respectively. This data is cropped away in the graphs to set an appropriate y axis for the rest of the data.
Type VIII lesions are also called "fibrotic lesions" or "fibrous plaques" [14]. These are most often present in the lower extremities [46]. Lipid pools are reduced or non-existent and the normal intima is replaced with fibrous connective tissue. The resorption of lipid is called a "regression of the lipid core". The Type VIII lesion is marked by disarrangement and death of the smooth muscle cells of the adjacent media. Adjacent media and adventitia contain macrophages, foam cells, and lymphocytes. [14]

[Caption] (Top row) The chemical structures of cholesterol and a triglyceride; from top to bottom on the trigylceride, the fatty acids are palmitic acid, oleic acid, alpha-linolenic acid (from Wikipedia). The glycerol (left piece of triglyceride), is the backbone of all triglyceride. (Second row) Esters of cholesterol formed via esterification with a fatty acid. (Third and fourth rows) Various forms of oxidized cholesterol, commonly found in athereosclerosis [47].
Cholesterol, \(\require{mhchem} \ce{C27H46O}\), is a hydrophobic, organic, lipid with three six member rings and one five member ring. It has 8 chiral centers and therefore 28 or 256 possible stereoisomers. Only one stereoisomer is synthesized in biological systems [48,49]. There is no "good" and "bad" cholesterol. There is only one cholesterol molecule. However, there are many esters of cholesterol and they are not all equally conducive to a long life. For example, there is some evidence cholesterol oleate is more atherogenic than cholesterol linoleate [50].
In atherosclerotic lesions, the concentration of oxidized cholesterol, or oxysterols, is at least 100 times greater than in plasma [51]. Oxysterols are thought to play an important role in triggering the immune response and in the transition from macrophage to foam cell. They are sometimes explored as a biomarker for atherosclerosis risk in research but as far as I can tell, plasma oxysterol concentrations are not a regular clinical lab test [47]. There is a review paper on oxysterols from the 1990s with a memorable quote "Increased understanding of this topic is bedeviled by the sheer number of published items, the lack of awareness of work published long ago, the redundancy and rediscovery seen in current work, repeated publication of abstracts without eventual full papers, dogmatic assertions of relevance to human health problems,and selective self-citation" [52]. As far as I can tell, the situation has improved little in the 25 years since.
While there may not be good and bad cholesterol, since cholesterol is a single molecule, there are more and less atherogenic cholesterol containing particles. Cholesterol is hydrophobic, to transport it in blood it must be water soluble so it is transported, along with fatty acids and triglycerides, in lipoproteins. All lipoproteins are not equally atherogenic.
A lipoprotein (commonly heard of as LDL, HDL, etc) is a 15-1500 nm diameter particle of lipids, cholesterol, and protein. The principle protein (without the other lipid/cholesterol molecules) responsible for the structure of the lipoprotein is the apolipoprotein [53]. Apo, the prefix, means a protein without a critical prosthetic group. For example, apohemoglobin is the hemoglobin protein without the heme, the iron containing molecule, normally bound to it [54].
Phospholipids are differentiated by the chemical structure of the heads. All the phospholipids in this paragraph are families defined by a common head group. The each member of the family usually has one or two hydrocarbon tails, 10-25 carbons long with 0-4 double bonds. Phosphatidylethanolamines, phosphatidylinositols, sphingomyelines are only transported by lipoproteins while phosphatidylcholine, lysophosphatidylcholine are present in free plasma and in lipoprotein particles. LDLs and VLDLs have relatively higher quantities of phosphatidylethanolamines and sphingomyelines. [55]
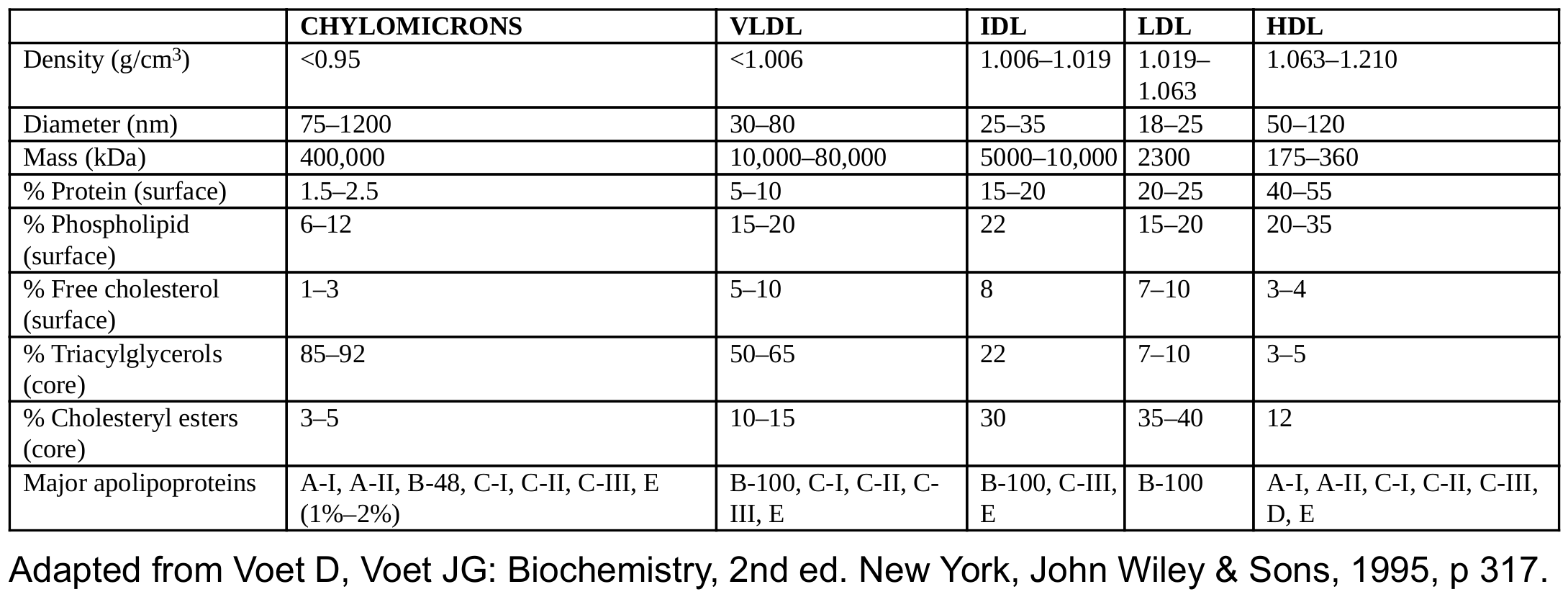
[Caption] Physical parameters of major lipoproteins [53].
Cholesterol particles are reported on blood tests as the serum concentration of cholesterol in mg/dL [56], ie, the mass of all the cholesterol inside the LDL particles divided by the volume of blood serum they came from is the LDL-cholesterol concentration.
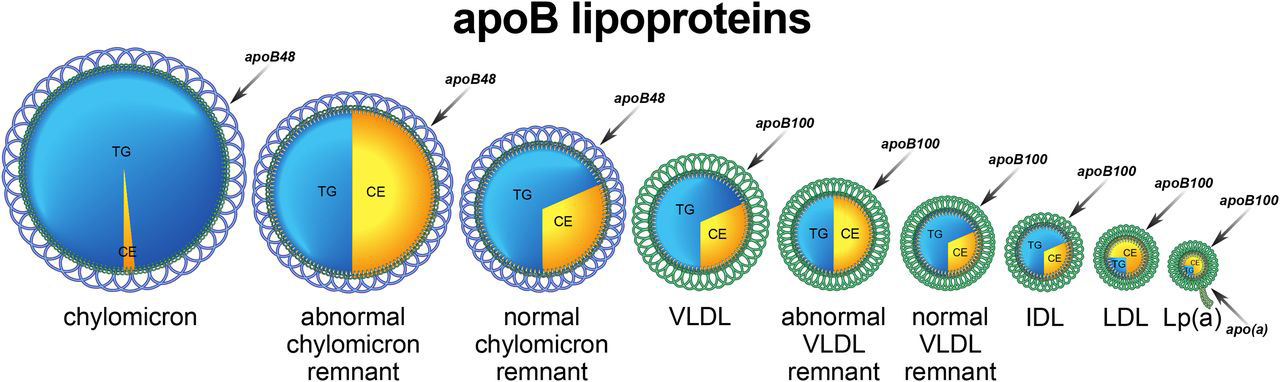
[Caption] Image from [57]. TG = triglycerides, CE = cholesterol esters. Not drawn anywhere close to scale.
LDL particles are heterogeneous in size and density, Each LDL particle contains exactly one apolipoprotein B (apoB, 4536 amino acids, single chain, 514 kDa). LDL transports trigylcerides, fatty acids, cholesterol, and cholesterol esters. Oxidation creates cholesterol esters (like cholesterol linoleate) from fatty acids in the particle (like linoleic acid) and cholesterol in an esterification [58,59]. I am confused because [60] seems to indicate about 200 apoB per LDL but that does not make sense at a number of levels. Need to understand better what is going on in that paper because authorities in the field are very clear that each LDL particle has exactly 1 apoB protein [56,61,62]. The often-cited paper which first showed apoB is exactly 1:1 with LDL particle number was published in 1988 [63], cannot find any revisions or retractions. Regardless of that misunderstanding, there are at least 100 proteins associated with LDL and VLDL particles including 15 apolipoproteins (A-I, A-II, A-IV, A-V, (a), B-100, B-48, C-I, C-II, C-III, C-IV, D, E, F, L1, and M) [60]. Some of the proteins identified have no obvious connection to lipid/cholesterol metabolism, like hemoglobin.
The size of LDL particles was hypothesized to affect atherogenesis but that seems to be disproven by the early 2000s [64]. Seems the main source of atherogenicity is number of particles.
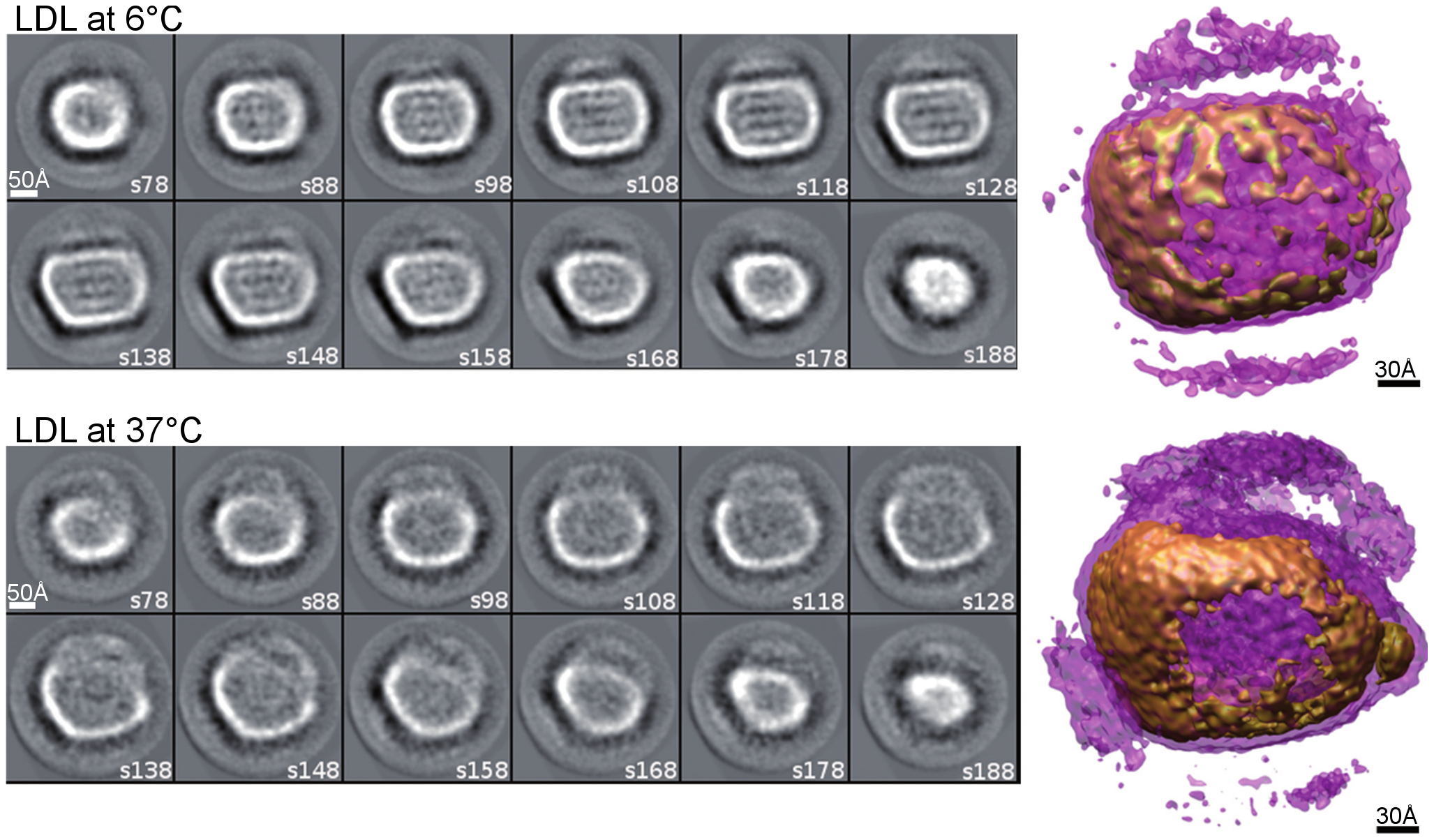
[Caption] Electron micrographs of LDL at different temperatures. Figure 3 from [65]. The purple surfaces on the models to the right indicate the lipid surface of the particles and the brown indicates the ApoB surface protein.
LDL discordance is defined as an unusual ratio of LDL-c to apoB. Normally apoB and LDL-c are correlated ("concordant apoB and LDL-c") where either both are elevated or depressed. Discordance with high apoB (and therefore low LDL-c) is caused by LDL particles having less cholesterol and a smaller average size [56]. In the case of discordance, it appears that the apoB number is more predictive of ASCVD outcomes than the LDL-c [56,66,67,68,69,70]. I only found one study indicating LDL-c was as useful as apoB/AI [71]. I was unable to find any evidence, cherry-picked, p-hacked, or otherwise, that LDL-c is a more significant risk factor than apoB (and therefore particle number) for ASCVD and downstream cardiovascular events. The plurality of the evidence suggests that reducing apo B or LDL-c earlier in life, decades before symptoms begin, is likely the best course of action for reducing ASCVD [72].

[Caption] Apo B vs LDL-c as a clinical predictor of long term outcomes [68,73].
The AMORIS study in Sweden (175,553 participants, 6 year follow up) showed apoB and the ratio of apoB to apo A-I were more predictive of fatal myocardial infarctions than triglycrides and total cholesterol [74]. These results have been reproducible: apoB is superior to non-HDL cholesterol (18,225 participants, 6 year follow up) [75]; "apo B and apo A-I are very strong predictors of atherosclerosis, whereas the lipid parameters are not" even after 1 year of statin therapy (6605 participants, 1 year follow up) [61,76] and reproduced (9,014 participants, 1 year follow up) [77]; apoB more predictive of MI than LDL cholesterol and triglycerides are independent factor (2,508 participants, 6 year follow up) [78].
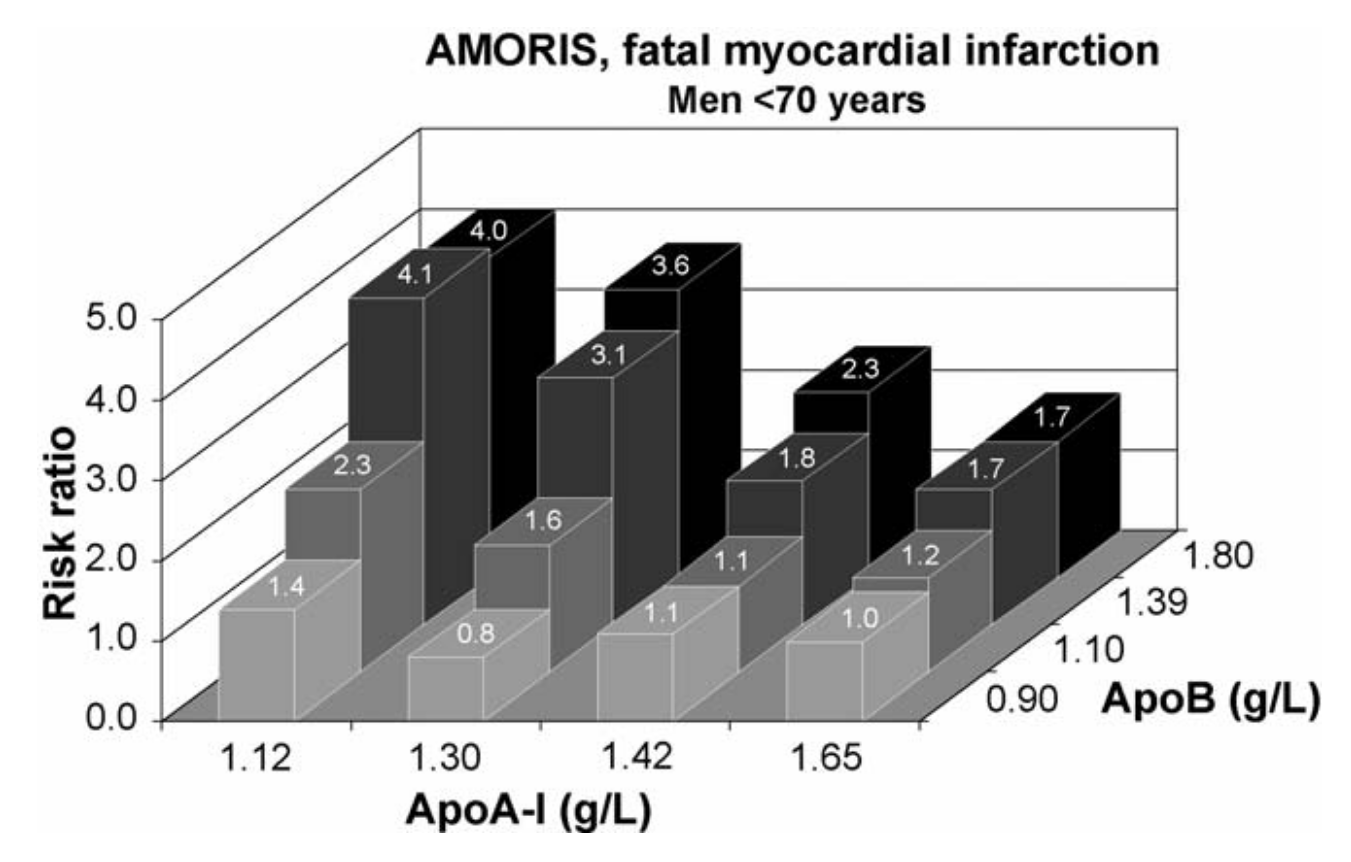
[Caption] From the AMORIS study [74].
Just as there are thousands of identified CYP proteins but only a handful have been tied to clinical utility, there are at least hundreds of lipoproteins but there is only evidence for the pathophysiological significance of a couple of them. Apolipoprotein A-I (or just apo A-I) is present on all HDL particles and sometimes in small amounts on LDL particles. Apolipoprotein B-100 (or commonly just apoB) is only associated with LDL, never found in HDL [61]. There is some literature surrounding Apo C-III and atherosclerosis. There is also conflicting information on which particles it is associated with. Several studies mention LDL apo C-III [61,79] in great detail but my physiology book indicates that no apolipoproteins except apoB are associated with LDL particles [80]. Physiology book is probably oversimplifying.
Lipoprotein(a) [LP(a), pronounced "L-P-little a" [56] to distinguish it from the apo A family] is an LDL particle containing the apolipoprotein (a) covalently bonded to the apoB. Because a subsection of the gene for LP(a) has a heritable and variable number of repeating subunits in a region called the kringle repeats, LP(a) comes in a variety of isoforms. Recent evidence suggests isoforms with less than 24 kringle repeats are more atherogenic [81] but it is unclear to me if the low kringle LP(a) particles are themselves more atherogenic or if the low kringle repeat isoform is associated with higher LP(a) concentrations [82]. Is it the apolipoprotein or is it the reduced concentration? The genetic variation of LP(a) is insanely complicated beyond just the kringle repeats [82]. High LP(a) is linked more to cardiovascular disease than to stroke [83].
LP(a) measurement is often discussed with confusing nomenclature. A 2013 review paper [84], confused LP(a) cholesterol with LP(a) mass [85]. LP(a) mass refers to the mass of the apolipoproteins, triglycrides, and cholesterol [86]. Or LP(a) mass refers to the mass of the apolipoproteins [87]. Or LP(a) mass refers to the mass of only the LP(a) apolipoprotein; it depends on the author [85]. As of 2019, this problem of definitions remains intractable [88] and the best a reader can do when looking into LP(a) is to be cognizant of the confusing inconsistencies.
Like LDL, HDL suffers from the confounding factor of HDL cholesterol concentration (HDL-c) vs HDL particle number (HDL-p) [89]. HDL-p is determined by NMR or ion mobility mass spec. However, again it seems to be the particle number, HDL-p, that is most important, not the HDL-c nor the apoAI (since HDL and apoAI do not have a 1:1 relationship like LDL and apoB) [90].
Because HDL particles are continuously adding and losing their lipid cargo, the HDL size distribution function for any individual is in a constant state of flux. There is evidence to suggest smaller HDL particles are more atheroprotective [91]. So with LDL you're better off with big particles, with HDL you want small ones.
Since at least 1951, it has been repeatedly observed that higher serum HDL cholesterol concentrations correlate with lower risk of cardiovascular disease and atherosclerosis [92,93,94]. However, pharmacological attempts to raise HDL-c have thus far not shown evidence of reduced ASCVD events and there have been several high-profile failures pursuing this strategy while successfully raising HDL-c [95]. Most of these drugs raise HDL-c by blocking the action of the cholesteryl ester transfer protein (they are CETP blockers), a protein that facilitates the movement of cholesterol esters between lipoproteins. Exactly how that increases HDL is beyond the scope of this article but like everything else, the more you learn, the more complicated it becomes [96].
The exact mechanism by which HDL exerts atheroprotective effects has yet to be conclusively shown though many theories exists. One of several things is true: {1} HDL-c levels rise with lower ASCVD due to so external cause driving both (therefore they are correlated but not causal). {2} The drugs available which raise HDL-c have undesirable side effects that cancel out the benefit of increased HDL-c. {3} The "protective effects" are dependent on the composition of HDL particles and the expression of various enzymes involved in HDL metabolism. {4} Residence times of HDL particles may play a role in the complication of HDL-c measurements and the failure of interventions with that target.
"Many studies have demonstrated that plasma LDL and HDL cholesterol and plasma total triglycerides are independent predictors of cardiovascular disease" [61,97]. Not sure if that means LDL cholesterol and HDL cholesterol and triglycerides (3 things) or if that means total cholesterol (sum of LDL and HDL) and triglycerides (2 things).
ApoE (in LDL and HDL) is highly atheroprotective as shown by the use of apoE deficient mice to study atherosclerosis [98].
Dyslipidemia has many definitions but generally means some combination of too much LDL-c, too much triglyceride, and/or too little HDL-c. It is defined as "the imbalance of lipids such as cholesterol, LDL-C, triglycerides, and HDL" [99]; or "elevated total or LDL cholesterol levels, or low levels of HDL cholesterol" [100]; or "increased levels of serum total cholesterol, LDL-C, triglycerides, or a decreased serum HDL-C concentration" [101].
Hypercholesterolemia is defined as "greater than 190 mg/dL, OR greater than 160 mg/dL with one major risk factor, OR greater than 130 mg/dL with two cardiovascular risk factors". Risk factors are age (over 45 and 55 for men and women, respectively), family history of ASCVD, hypertension, diabetes, smoking, and low HDL-c (40 and 55 mg/dL for men and women, respectively) [102].
PCSK9 is a serine protease which degrades LDL receptors (mostly found in hepatocytes). PCSK9 is a drug target because inhibition of PCSK9 increases the number of LDL receptors and thereby increases the clearance rate of LDL [103]. PCSK9 gain of function mutations are associated with high LDL/apoB concentrations while PCSK9 loss of function mutations are associated with sometimes dramatic reductions in LDL-c (80% decrease from normal). These loss of function mutations are rare enough that the consequences are documented in case reports rather than long term studies but there is no evidence yet they cause harm [104]. The only study long term study I found was a 15 year follow up on 87 black subjects and 305 white subjects which did show a significant and expected reduction in coronary heart disease [105]. However, despite that being a small sample study, no other evidence has come to light showing harms from extremely low LDL-c caused by naturally occuring PCSK9 loss of function mutations (that I can find).
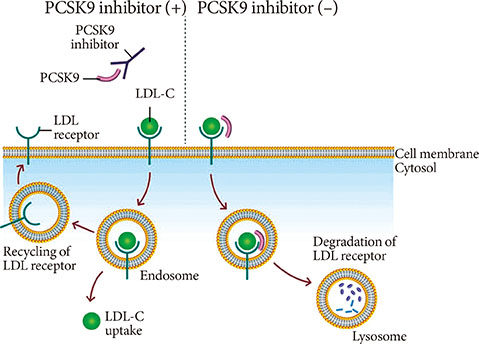
[Caption] Mechanism of PCSK9 and PCSK9 inhibitors, taken from [106].
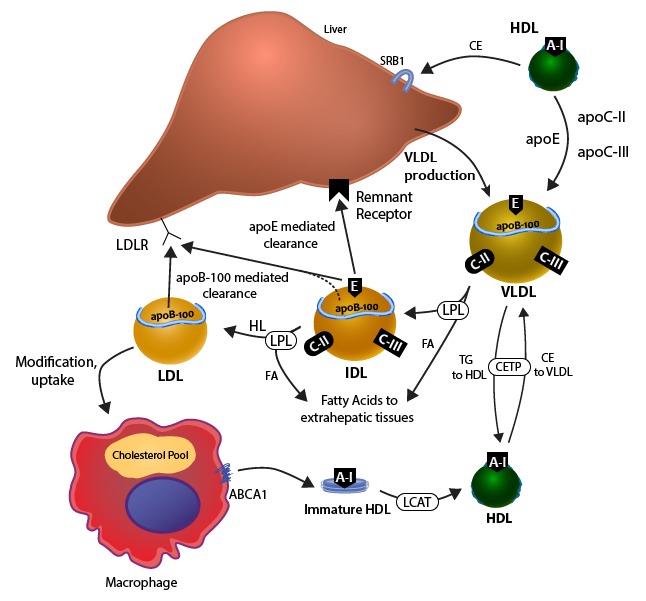
[Caption] Metabolism of apoB 100 lipoproteins from [28]. Of the players on this page, most have been the target of a pharmaceutical intervention. CETP [107] which was not successful at all [108,109], LDLR (through PCSK9 inhibition) [110,111], LPL [112,113] (that fibrates target LPL is unclear but often suggested), and ABCA1 [114,115].
The microsomal triglyceride transport protein (MTP) assembles the LDL particles in the hepatocytes [116]. Abetalipoproteinemia is a genetic deficency in MTP which requires medical intervention [117]. The condition is too rare to properly assess its effect on ASCVD risk [118] but it has other undesirable effects.
Nitric oxide, \(\require{mhchem} \ce{NO}\), is a paracrine signaling molecule of the endothelium inducing the relaxation of smooth muscle cells. As a strong oxidizing agent, overproduction of \(\require{mhchem} \ce{NO}\) is atherogenic [21,119]. Oxidizing of cholesterol into cholesterol esters is likely a key step of atheropathogenesis.
Lipemic plasma refers to plasma with an abnormally high level of lipid. Normal plasma is 8% lipid/92% aqueous; lipemic plasma is up to 25% lipid. There is no fixed distribution of particles where this lipid resides (i.e. the ratios of chylomicrons:HDL:LDL:VDL are not fixed). Obviously the turbidity of the sample is higher when the lipid is in larger particles. Common figures range from 50% in VLDL to 90% chylomicrons. [120]
As noted above, triglycerides are usually regarded as an independent risk factor for ASCVD. Details to be researched later.
The following are currently used treatments for dyslipidemia/hypercholesterolemia:
Statins: target cholesterol synthesis; lowers LDL-c and triglycerides; slightly raises HDL-c.
CETP inhibitors: raise HDL-c but largely failed in clinical trials.
Ezetimibe: cholesterol absorption inhibitor; lowers LDL-c and triglycerides; slightly raises HDL-c.
PCSK9 inhibitors: decreases LDL-c. Costly, reserved high risk people who are intolerance or unresponsive to statins.
Citrate lyase inhibitors: decreases LDL-c.
Bile acid sequestrants: lowers LDL-c; slightly raises HDL-c.
Fibrates: lowers LDL-c, increases HDL-c, slightly lowers triglycerides
Niacin: lowers LDL-c and triglycerides; raises HDL-c.
Omega-3 fatty acids: decreases triglycerides
The following sections will discuss all these treatments and lot more that did not make it through drug trials.
Torcetrapib was a Pfizer drug which successfully lowered HDL-c. However, side effects made the treatment worse than the ailment (increases in blood pressure [121,122], changes in electrolyte balance [123]). The big Torcetrapib trail was abandoned for patient safety reasons in 2006 [124].
Dalcetrapib, from pharmaceutical giant Roche, also raised HDL-c but failed to improve outcomes [125].
Evacetrapib, from Eli-Lilly, followed the same story. It "produced an increase in HDL-c up to 128% and a 35% decrease in LDL-c, in comparison to placebo ... more potent than previous CETP inhibitors" but "evacetrapib’s phase III ACCELERATE trial showed no significant reduction in major adverse cardiovascular events or mortality, and the drug will not be marketed" [107].
Anacetrapib, from Merck, shows some evidence of efficacy but this success is likely the result of reducing non-HDL cholesterol rather than by increasing HDL [126].
ApoA1 mimetic peptides are also an ongoing field of research targeting HDL. I cannot find any examples of drugs that have been brought to market with this mechanism but neither can I find evidence this area of research should be abandoned [127].
The hypothesis that higher HDL-c is causally protective against athereosclerosis is contradicted by the fact genetic mutations which cause high HDL-c are not themselves protective against ASCVD [128]. Importantly, there are mutations that cause high HDL-p, smaller HDLs, and are associated with lower CVD incidences [129]. To me the literature indicates the field is pivoting toward the study of particle size in HDL.
Mipomersen, from Genzyme Corp, is an antisense nucleotide that binds the apoB messenger RNA and prevents transcription. This targets the synthesis of apoB and therefore prevents the assembly of apoB particles [130]. As of 2013, it is currently only available for homozygous familial hypercholesterolemia [131].
There is no doubt that statins are effective in lowering apoB, LDL-c, and non-HDL-c (see Table 1 in [132] for seven large scale study examples). Atorvastatin, fluvastatin, lovastatin, pravastatin, pitavastatin, rosuvastatin, simvastatin are the seven currently approved in the US.
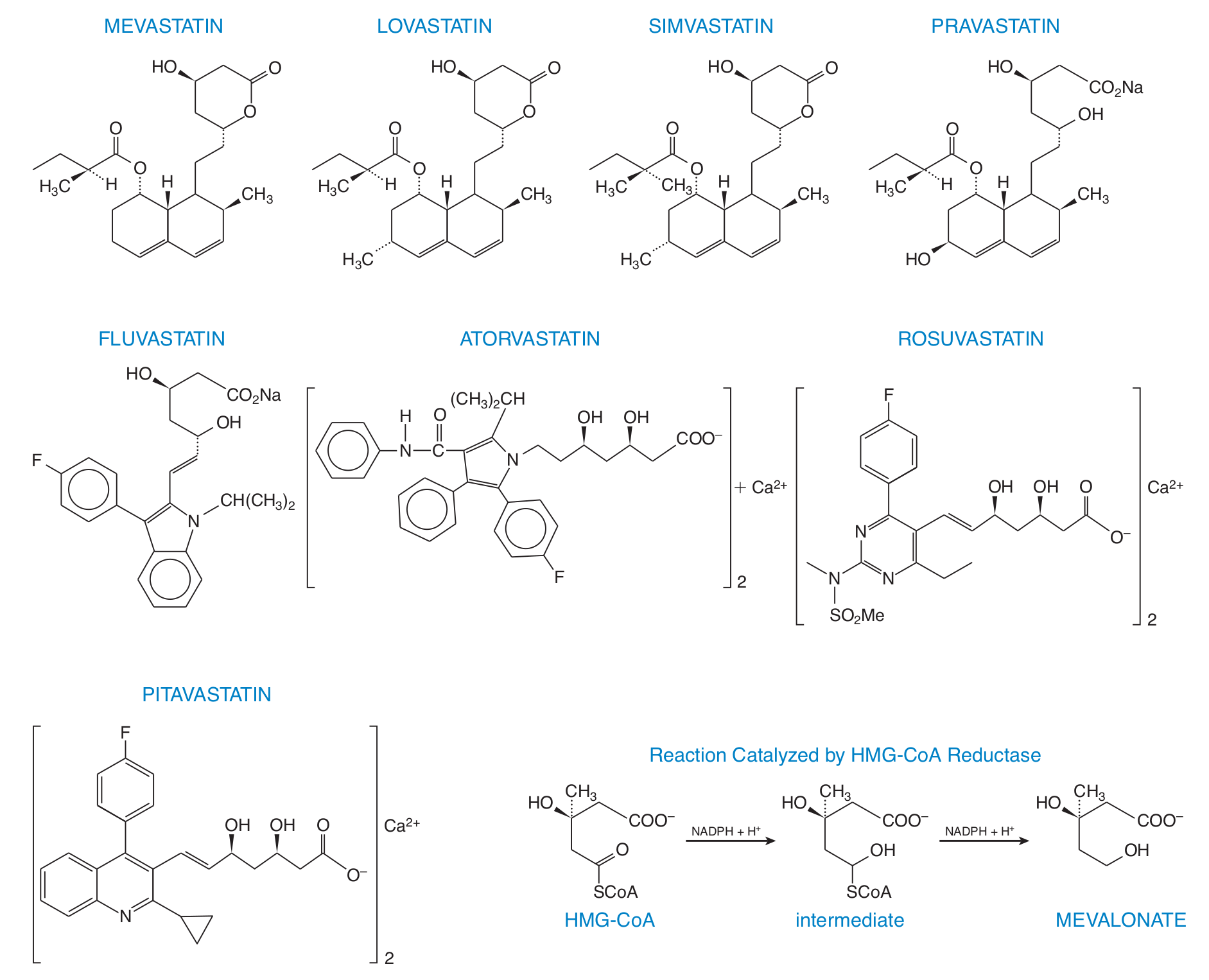
[Caption] The structures of the common statins and the rate limiting step of cholesterol synthesis they target, taken from [133].
Statins were first isolated from mold and identified as cholesterol synthesis inhibitors in 1976 [134]. Lovastatin (formerly mevinolin) was the first to market in the 1980's [135]. Statins are well-studied and well-tolerated drugs across populations [136,137]. Statins can be hepatotoxic and slightly elevate the risk of rhabdomyolysis, diabetes [133,138], and hemorrhagic stroke [139]. Suspected side effects of statins that are not proven but still the subject of study include elevated cancer risk, peripheral neuropathy, mood disorders, interstitial lung disease, and acute interstitial nephritis [138]. Every drug ever prescribed this broadly for this long has a list of "possible" side effects at least as long.
As more data is collected, support for starting statin therapy early is increasing [140,141,142].
Ezetimibe is most often discussed in combination with statin therapy for those who do not meet LDL-c targets on statins alone [143,144,145]. It inhibits a cholesterol transporter protein in the jejunal brush border, the Nieman Pick C1 Like 1 protein [146]. It does not seem to be terribly effective as evidence by the fact it is so often used as the non-placebo control for studies of other lipid drugs [my interpretation| value?].
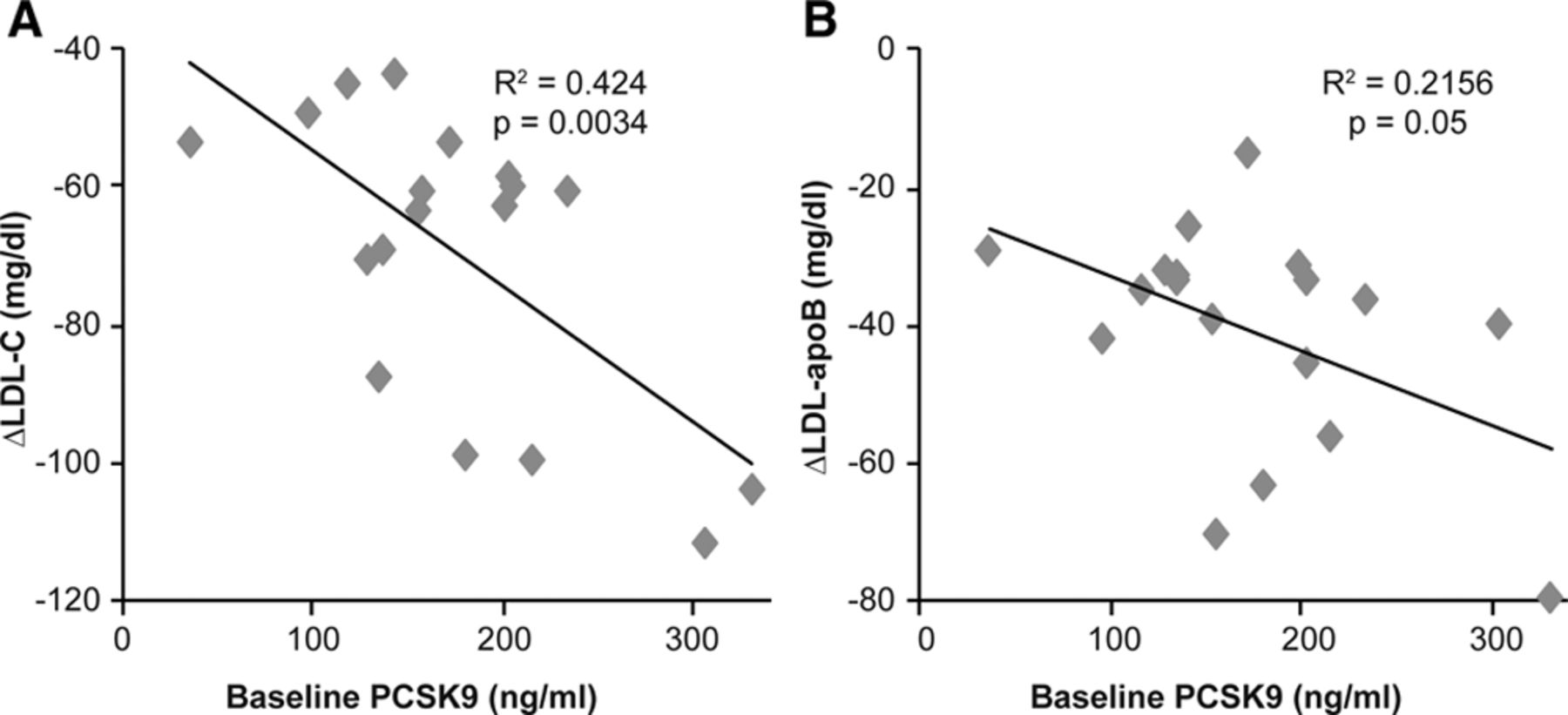
[Caption] PCSK9 inhibitor (alirocumab) successfully lowers LDL-c and apoB in healthy humans [147].
Currently alirocumab and evolocumab are monoclonal antibodies targeting PCSK9. Frustratingly, studies and reviews consistently point to LDL-c metrics instead of apoB [148] though there are some reanalyses that focuses on apoB [149]. PCSK9 inhibitors began phase 3 clinical trials around 2015 with high anticipation [150,151]. The results have been impressive reducing apoB levels from 80-100 mg/dL to under 50 [149]; multiple studies have shown 40-60% reduction in apoB [152]. Reviews of PCSK9 inhibitors universally support them as efficacious in lowering LDL/apoB [149,153,154] but only a few mentioned positive effects on mortality and cardiovascular events [155,156,157]. These finding are not without controversy. This review suggests that evolocumab increased mortality in clinical trials [158].
At over $12,000 per year, the primary known drawback to PCSK9 inhibitors is the cost benefit trade-off [159]. In the general population, people are willing to pay at most $50,000 per quality adjusted year of life [160]. When considering time-discounted cash flows, paying $12,000 per year at age 20 to prevent adverse events at age 60 becomes untenable. However, over time as competition continues and patents expire, we expect the cost come down.
I heard of a PCSK9 trial that ended the control arm prematurely because the efficacy of the drugs were so great that continuing the control arm became unethical. I have not been able to find a news article or journal article documenting that occurrence.

[Caption] The structures of the three bile acid sequestrants, taken from [133].
Bile acid sequestrants are long polymers which do not absorb from the GI system. They are highly positively charged so they bind to the negatively charged bile acids and are then excreted. Generally well tolerated and safe as they do not enter the body; however there can be GI side effects [133].

[Caption] The structures of the three bile acid sequestrants, taken from [133].
The mechanism by which fibrates lower LDL-c or raise HDL-c remains unclear [133] though it is usually claimed to activate lipoprotein lipase (LPL) [113] or by activating PPRE genes through PPARα signaling [161]. Because they were largely ineffective at lowering major adverse cardiac events, fibrates have fallen out of favor [133]. Safety concerns, including Steven Johnson syndrome [162], probably contributed to their declining popularity.

[Caption] Niacin structure from Wikipedia.
Niacin failed to improve clinical outcomes "despite significant improvements in HDL cholesterol and triglyceride levels" [163].
The only drug in this category is bempedoic acid but it is so obscure that Goodman and Gilman's has no mention of it [164]. I only mention it because it appears on the Mayo clinic website as an LDL lowering drug. If it is too obscure for Goodman's, it is too obscure for my article.
I was confused by the results on dietary cholesterol. We originally learned that cholesterol played an important role in atherosclerosis from Anichkov's experiments feeding cholesterol to rabbits [8,9]. However, it is easy to find studies demonstrating no link between atherosclerosis and dietary cholesterol. No link between dietary cholesterol and subclinical arterial calcification in Korean adults [165]; weak association between dietary cholesterol and LDL-c [166]; dietary cholesterol increases LD-c and HDL-c in young healthy women [167]; reducing saturated fat intake increases LDL receptors [168]; etc. It is also just as easy to find studies questioning the link between dietary cholesterol and atherogenic burden [169,170,171].
The explanatory factor is that there is tremendous genetic variation in the individual responses to dietary cholesterol [172]. Part of that variation can be traced back to the Nieman Pick C1 Like 1 protein [173]. As of 2013 (and, to the best I can tell, today) there is no easy way to distinguish hypo vs hyper responders without testing a dietary intervention [174].
This is all the stuff that did not quite fit but I found helpful in the process. The immune response did not seem to be much of a drug target for ASCVD prevention and the calcification process does not seem to be a drug target at all.
Remnant type III hyperlipoproteinemia is a genetic disorder which cannot be diagnosed without an apoB lab measurement. It is characterized by high cholesterol, low apoB, and high triglyceride. The LDL becomes extremely cholesterol rich and the condition is highly atherogenic [175].

[Caption] From Wikipedia, a summary of hematopoiesis similar to the textbooks [176,177]. I added the foam cell for clarity.
In the development of atherosclerosis, the mast cells, T-cells, macrophages, and foam cells are the dominant players. Later, when a lesion becomes "complicated", i.e. Type VI, thrombocytes do the work of forming a thrombus or hematoma. Apo C-III induces monocyte attachment to endothelium [178]. Monocytes are precursor cells to macrophages and foam cells.
Osteoclasts [179,180,181] and osteoblasts [182,183] are involved in the calcification of atheromas (i.e. the transition to Type VII). In healthy bones, osteoclasts continuously dissolve bone, osteoblasts continually create bone, and osteocytes send paracrine signals to stimulate/inhibit osteoclasts and osteoblasts [80].
As I was scribbling this down, I thought the immune response and calcification would be more important. Available therapies (that I've read up on) almost exclusively target triglycerides and cholesterol in the blood rather than attacking the mechanisms of calcification and inflammation.
[1] "Blood Vessels," in Pathologic basis of disease, 9th Ed, pp. 491—501, 2015.
[2] On the insensitivity and irritability of certain parts of animals - translated from Italian by Google, 1755.
[3] "Albrecht von Haller," in Pioneers in Pathology, pp. 519—522, 2017.
[4] "Lecture XV: Passive Processes. Fatty Degeneration," in Lectures on cellular pathology: physiological and pathological histology, pp. 316—341, 1858.
[5] "Rudolf Virchow," in Pioneers in Pathology, pp. 514—517, 2017.
[6] "Ueber Atherosclerosis," Verhandlungen Der Kongresse Fuer Innere Medizin, vol. 21, 1904.
[7] "Nikolai N. Anichkov and his theory of atherosclerosis," Texas Heart Institute Journal, vol. 33, pp. 417—423, 2006.
[8] "Uber experimentelle Cholesterinsteatose und ihre Bedentung fiir die Entstehung einiger pathologischer Prozesse," Ebl. Allg. Path. Path. Anat, vol. 24, 1913.
[9] "Classics in arteriosclerosis research: On experimental cholesterin steatosis and its significance in the origin of some pathological processes," Arteriosclerosis, vol. 3, pp. 178—182, 1983.
[10] "Cardiology's 10 greatest discoveries of the 20th century," Texas Heart Institute Journal, vol. 29, pp. 164, 2002.
[11] "In celebration of the 100th anniversary of the lipid hypothesis of atherosclerosis," Journal of Lipid Research, vol. 54, pp. 2946—2949, 2013.
[12] "Nikolaj Anichkov," in Pioneers in Pathology, pp. 23—27, 2017.
[13] Lectures on Pathology, 1924.
[14] "A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association," Circulation, vol. 92, pp. 1355—1374, 1995.
[15] "The Deadly Disease," in Chlamydia Atherosclerosis Lesion: Discovery, Diagnosis and Treatment, 2007.
[16] "Atherosclerosis: a journey around the terminology," in Atherosclerosis, Arteriosclerosis and Arteriolosclerosis, 2020.
[17] "Historical review of research on atherosclerosis," Nutrition and Biotechnology in Heart Disease and Cancer, pp. 1—6, 1995.
[18] "Arteriosclerosis: rethinking the current classification," Archives of Pathology & Laboratory Medicine, vol. 133, pp. 1309—1316, 2009.
[19] "Gabriel Fallopius," Encyclopedia Britannica, 1999.
[20] Lectiones Gabrielis Fallopii De Partibus Similaribus Humani Corporis, 1575.
[21] "The pathogenesis of atherosclerosis," Medicine, vol. 42, pp. 480—484, 2014.
[22] "Pathology of Vulnerability Caused by High-Risk (Vulnerable) Arteries and Plaques," in Asymptomatic Atherosclerosis, pp. 39—51, 2011.
[23] "The Circulatory System," in Basic Histology Text and Atlas, pp. 220—222, 2016.
[24] "Measurement of aortic intimal-medial thickness in adolescents and young adults," Ultrasound in Medicine & Biology, vol. 36, pp. 560—565, 2010.
[25] "Circulatory System," in Functional Histology Text and Color Atlas, pp. 149, 2014.
[26] "Natural history and histological classification of atherosclerotic lesions: an update," Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 20, pp. 1177—1178, 2000.
[27] "AHA Medical Scientific Statement: A Definition of Initial, Fatty Streak, and Intermediate Lesions of Atherosclerosis," Circulation, vol. 89, pp. 2462—2478, 1994.
[28] "The role of lipids and lipoproteins in atherosclerosis," Endotext, 2019.
[29] "Grading atherosclerosis in aorta and coronary arteries obtained at autopsy: application of a tested method," Bulletin of the World Health Organization, vol. 31, pp. 297, 1964.
[30] "The presence of lipid droplets and the absence of stable sudanophilia in osmium-fixed human leukocytes," Journal of Histochemistry & Cytochemistry, vol. 19, pp. 551—557, 1971.
[31] "Stable sudanophilia of human neutrophil leucocytes in relation to peroxidase and oxidase," Journal of Histochemistry & Cytochemistry, vol. 1, pp. 8—26, 1953.
[32] "Influence of the geometry of the left main coronary artery bifurcation on the distribution of sudanophilia in the daughter vessels," Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 17, pp. 1356—1360, 1997.
[33] "Imaging of lipids in atheroma by desorption electrospray ionization mass spectrometry," Analytical Chemistry, vol. 81, pp. 8702—8707, 2009.
[34] "Comparative lipidomics profiling of human atherosclerotic plaques," Circulation: Cardiovascular Genetics, vol. 4, pp. 232—242, 2011.
[35] "Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults," Arteriosclerosis, vol. 9, pp. I19—32, 1989.
[36] "Compensatory enlargement of human atherosclerotic coronary arteries," New England Journal of Medicine, vol. 316, pp. 1371—1375, 1987.
[37] "Stenting-induced Vasa Vasorum compression and subsequent flow resistance: a finite element study," Biomechanics and Modeling in Mechanobiology, vol. 20, pp. 121—133, 2021.
[38] "Pathology of Vulnerability Caused by High-Risk (Vulnerable) Arteries and Plaques," in pp. 39—51, 2011.
[39] "Atherosclerotic plaque volume and composition in symptomatic carotid arteries assessed with multidetector CT angiography; relationship with severity of stenosis and cardiovascular risk factors," European Radiology, vol. 19, pp. 2294—2301, 2009.
[40] "Regulatory mechanisms in vascular calcification," Nature Reviews Cardiology, vol. 7, pp. 528—536, 2010.
[41] "Crystal structure of hydroxyapatite," Nature, vol. 204, pp. 1050—1052, 1964.
[42] "Coronary artery calcium: a multi-institutional, multimanufacturer international standard for quantification at cardiac CT," Radiology, vol. 243, pp. 527—538, 2007.
[43] "Quantification of coronary artery calcium using ultrafast computed tomography," Journal of the American College of Cardiology, vol. 15, pp. 827—832, 1990.
[44] "High dose and long-term statin therapy accelerate coronary artery calcification," International Journal of Cardiology, vol. 184, pp. 581—586, 2015.
[45] "Distribution of coronary artery calcium by race, gender, and age: results from the Multi-Ethnic Study of Atherosclerosis (MESA)," Circulation, vol. 113, pp. 30—37, 2006.
[46] "Human atherosclerosis. I. Cell constitution and characteristics of advanced lesions of the superficial femoral artery," The American Journal of Pathology, vol. 114, pp. 79, 1984.
[47] "Macrophage oxysterols and their binding proteins: roles in atherosclerosis," Current Opinion in Lipidology, vol. 23, pp. 462—470, 2012.
[48] "Chiral aggregation phenomena. 4. A search for stereospecific interactions between highly purified enantiomeric and racemic dipalmitoyl phosphatidylcholines and other chiral surfactants in monolayers, vesicles, and gels," Journal of the American Chemical Society, vol. 104, pp. 636—639, 1982.
[49] "Enantiospecificity of cholesterol function in vivo," Journal of Biological Chemistry, vol. 276, pp. 44369—44372, 2001.
[50] "LDL cholesteryl oleate as a predictor for atherosclerosis: evidence from human and animal studies on dietary fat," Journal of Lipid Research, vol. 50, pp. S434—S439, 2009.
[51] "Oxysterols and atherosclerosis," Atherosclerosis, vol. 142, pp. 1—28, 1999.
[52] "Review of progress in sterol oxidations: 1987—1995," Lipids, vol. 31, pp. 453—487, 1996.
[53] "Hepatobiliary function," in Medical Physiology, pp. 967, 2017.
[54] "The kinetic mechanism of heme binding to human apohemoglobin.," Journal of Biological Chemistry, vol. 258, pp. 4298—4303, 1983.
[55] "A phospholipidomic analysis of all defined human plasma lipoproteins," Scientific Reports, vol. 1, pp. 1—11, 2011.
[56] "Episode 3: A deep dive into heart disease," The Drive Podcast, 2018.
[57] "Hypertriglyceridemia and cardiovascular risk: a cautionary note about metabolic confounding," Journal of Lipid Research, vol. 59, pp. 1266—1275, 2018.
[58] "Structure of apolipoprotein B-100 in low density lipoproteins," Journal of Lipid Research, vol. 42, pp. 1346—1367, 2001.
[59] "LDL particle size distribution. Results from the Framingham Offspring Study," Arteriosclerosis and Thrombosis: A Journal of Vascular Biology, vol. 12, pp. 1410—1419, 1992.
[60] "Proteome of human plasma very low-density lipoprotein and low-density lipoprotein exhibits a link with coagulation and lipid metabolism," Thrombosis and Haemostasis, vol. 112, pp. 518—530, 2014.
[61] "The apolipoprotein story," Atherosclerosis Supplements, vol. 7, pp. 23—27, 2006.
[62] "ApoB: the power of physiology to transform the prevention of cardiovascular disease," Circulation Research, vol. 124, pp. 1425—1427, 2019.
[63] "Plasma very low density lipoproteins contain a single molecule of apolipoprotein B," Journal of Lipid Research, vol. 29, pp. 1461—1473, 1988.
[64] "Low-density lipoprotein size and cardiovascular disease: a reappraisal," The Journal of Clinical Endocrinology & Metabolism, vol. 88, pp. 4525—4532, 2003.
[65] "Three-dimensional cryoEM reconstruction of native LDL particles to 16 Å resolution at physiological body temperature," PloS One, vol. 6, pp. e18841, 2011.
[66] "Discordance between apolipoprotein B and LDL-cholesterol in young adults predicts coronary artery calcification: the CARDIA study," Journal of the American College of Cardiology, vol. 67, pp. 193—201, 2016.
[67] "Clinical implications of discordance between low-density lipoprotein cholesterol and particle number," Journal of Clinical Lipidology, vol. 5, pp. 105—113, 2011.
[68] "Discordance of low-density lipoprotein (LDL) cholesterol with alternative LDL-related measures and future coronary events," Circulation, vol. 129, pp. 553—561, 2014.
[69] "Discordance analysis and the Gordian Knot of LDL and non-HDL cholesterol versus apoB," Current Opinion in Lipidology, vol. 25, pp. 461—467, 2014.
[70] "ApoB: the power of physiology to transform the prevention of cardiovascular disease," Circulation Research, vol. 124, pp. 1425—1427, 2019.
[71] "Prognostic utility of ApoB/AI, total cholesterol/HDL, non-HDL cholesterol, or hs-CRP as predictors of clinical risk in patients receiving statin therapy after acute coronary syndromes: results from PROVE IT—TIMI 22," Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 29, pp. 424—430, 2009.
[72] "Eradicating the burden of atherosclerotic cardiovascular disease by lowering apolipoprotein B lipoproteins earlier in life," Journal of the American Heart Association, vol. 7, pp. e009778, 2018.
[73] "LDL cholesterol: controversies and future therapeutic directions," The Lancet, vol. 384, pp. 607—617, 2014.
[74] "High apolipoprotein B, low apolipoprotein AI, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study," The Lancet, vol. 358, pp. 2026—2033, 2001.
[75] "Non-HDL cholesterol and apolipoprotein B in the prediction of coronary heart disease in men," Circulation, vol. 112, pp. 3375—3383, 2005.
[76] "Relation between baseline and on-treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS)," Circulation, vol. 101, pp. 477—484, 2000.
[77] "Relationship between lipid levels and clinical outcomes in the Long-term Intervention with Pravastatin in Ischemic Disease (LIPID) Trial: to what extent is the reduction in coronary events with pravastatin explained by on-study lipid levels?" Circulation, vol. 105, pp. 1162—1169, 2002.
[78] "Nonfasting apolipoprotein B and triglyceride levels as a useful predictor of coronary heart disease risk in middle-aged UK men," Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 22, pp. 1918—1923, 2002.
[79] "LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients," Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 23, pp. 853—858, 2003.
[80] "The Parathyroid Glands and Vitamin D," in Medical Physiology, pp. 3350, 2017.
[81] "First Demonstration That Small Lp (a) Particles, With Kringle IV Repeats of 24 or Less, but Not Large Lp (a) Particles (More Than 24 Kringle IV Repeats), Are Atherogenic," Circulation, vol. 138, pp. A16589—A16589, 2018.
[82] "Lipoprotein (a) beyond the kringle IV repeat polymorphism: the complexity of genetic variation in the LPA gene," Atherosclerosis, vol. 349, pp. 17—35, 2022.
[83] "Lipoprotein (a) as a cardiovascular risk factor: current status," European Heart Journal, vol. 31, pp. 2844—2853, 2010.
[84] "Lipoprotein (a), cardiovascular disease, and contemporary management," vol. 88, pp. 1294—1311, 2013.
[85] "Lipoprotein (a) mass: a massively misunderstood metric," Journal of Clinical Lipidology, vol. 8, pp. 550—553, 2014.
[86] "Immunochemical quantification of human plasma Lp (a) lipoprotein," Lipids, vol. 9, pp. 15—26, 1974.
[87] "Electrophoretic measurement of lipoprotein (a) cholesterol in plasma with and without ultracentrifugation: comparison with an immunoturbidimetric lipoprotein (a) method," Clinical Biochemistry, vol. 37, pp. 481—488, 2004.
[88] "The challenges of measuring Lp (a): A fight against Hydra?" Atherosclerosis, vol. 289, pp. 181—183, 2019.
[89] "HDL-C vs HDL-P: how changing one letter could make a difference in understanding the role of high-density lipoprotein in disease," U.S. Patent 11, 2014.
[90] "High-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy," Circulation, vol. 128, pp. 1189—1197, 2013.
[91] "Biological activities of HDL subpopulations and their relevance to cardiovascular disease," Trends in Molecular Medicine, vol. 17, pp. 594—603, 2011.
[92] "Protein-lipid relationships in human plasma: II. In atherosclerosis and related conditions," The American Journal of Medicine, vol. 11, pp. 480—493, 1951.
[93] "The complexity of HDL," Biochimica Et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids, vol. 1801, pp. 1286—1293, 2010.
[94] "High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target," Circulation Research, vol. 114, pp. 171—182, 2014.
[95] "HDL hypothesis: where do we stand now?" Current Atherosclerosis Reports, vol. 16, pp. 1—9, 2014.
[96] "Separating the mechanism-based and off-target actions of cholesteryl ester transfer protein inhibitors with CETP gene polymorphisms," Circulation, vol. 121, pp. 52—62, 2010.
[97] "Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III)," JAMA, vol. 285, pp. 2486—2497, 2001.
[98] "Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells," Cell, vol. 71, pp. 343—353, 1992.
[99] "Dyslipidemia," in StatPearls, 2022.
[100] "Primary prevention of CVD: treating dyslipidemia," American Family Physician, vol. 83, pp. 1207—1208, 2011.
[101] "Dyslipidemia and cardiovascular disease risk among the MASHAD study population," Lipids in Health and Disease, vol. 19, pp. 1—11, 2020.
[102] "Hypercholesterolemia," in StatPearls, 2022.
[103] "Molecular biology of PCSK9: its role in LDL metabolism," Trends in Biochemical Sciences, vol. 32, pp. 71—77, 2007.
[104] "Living the PCSK9 adventure: from the identification of a new gene in familial hypercholesterolemia towards a potential new class of anticholesterol drugs," Current Atherosclerosis Reports, vol. 16, pp. 1—23, 2014.
[105] "Sequence variations in PCSK9, low LDL, and protection against coronary heart disease," New England Journal of Medicine, vol. 354, pp. 1264—1272, 2006.
[106] "New drugs for treating dyslipidemia: beyond statins," Diabetes & Metabolism Journal, vol. 39, pp. 87—94, 2015.
[107] "Evacetrapib: another CETP inhibitor for dyslipidemia with no clinical benefit," Cardiology in Review, vol. 25, pp. 43—52, 2017.
[108] "Trials and tribulations of CETP inhibitors," Circulation Research, vol. 122, pp. 106—112, 2018.
[109] "CETP inhibition: past failures and future hopes," Clinical Medicine Insights: Cardiology, vol. 10, pp. CMC—S32667, 2016.
[110] "An update on PCSK9 inhibitors-pharmacokinetics, drug interactions, and toxicity," Expert Opinion On Drug Metabolism & Toxicology, vol. 16, pp. 1199—1205, 2020.
[111] "PCSK9: regulation and target for drug development for dyslipidemia," Annual Review of Pharmacology and Toxicology, vol. 57, pp. 223—244, 2017.
[112] "An anti-ANGPTL3/8 antibody decreases circulating triglycerides by binding to a LPL-inhibitory leucine zipper-like motif," Journal of Lipid Research, vol. 63, 2022.
[113] "Mechanism of action of fibrates," Postgraduate Medical Journal, vol. 69, pp. S34—41, 1993.
[114] "ABCA1 as a New Therapeutic Target for Treating Cardiovascular Disease," Drug News & Perspectives, vol. 15, pp. 24—28, 2002.
[115] "SIRT1 activator E1231 protects from experimental atherosclerosis and lowers plasma cholesterol and triglycerides by enhancing ABCA1 expression," Atherosclerosis, vol. 274, pp. 172—181, 2018.
[116] "Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia," Science, vol. 258, pp. 999—1001, 1992.
[117] Abetalipoproteinemia, 2022.
[118] "Clinical implications of lipid genetics for cardiovascular disease," Current Cardiovascular Risk Reports, vol. 4, pp. 461—468, 2010.
[119] "Vascular endothelium in atherosclerosis," Cell and Tissue Research, vol. 335, pp. 191—203, 2009.
[120] "Lipemia: causes, interference mechanisms, detection and management," Biochemia Medica, vol. 24, pp. 57—67, 2014.
[121] "Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone," British Journal of Pharmacology, vol. 154, pp. 1465—1473, 2008.
[122] "Effects of CP-532,623 and torcetrapib, cholesteryl ester transfer protein inhibitors, on arterial blood pressure," Journal of Cardiovascular Pharmacology, vol. 53, pp. 507—516, 2009.
[123] "Aldosterone, sodium, and hypertension: lessons from torcetrapib?" Hypertension, vol. 55, pp. 221—223, 2010.
[124] "Pfizer stops clinical trials of heart drug," 2006.
[125] "Effects of dalcetrapib in patients with a recent acute coronary syndrome," New England Journal of Medicine, vol. 367, pp. 2089—2099, 2012.
[126] "Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin," European Heart Journal, vol. 36, pp. 39—50, 2015.
[127] "Apolipoprotein mimetic peptides: an emerging therapy against diabetic inflammation and dyslipidemia," Biomolecules, vol. 11, pp. 627, 2021.
[128] "Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study," The Lancet, vol. 380, pp. 572—580, 2012.
[129] "Genetic variation at the phospholipid transfer protein locus affects its activity and high-density lipoprotein size and is a novel marker of cardiovascular disease susceptibility," Circulation, vol. 122, pp. 470—477, 2010.
[130] "Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia," Circulation, vol. 129, pp. 1022—1032, 2014.
[131] "FDA approves new orphan drug Kynamro to treat inherited cholesterol disorder," 2013.
[132] "Relations of change in plasma levels of LDL-C, non-HDL-C and apoB with risk reduction from statin therapy: a meta-analysis of randomized trials," Journal of the American Heart Association, vol. 3, pp. e000759, 2014.
[133] "Drug Therapy for Hypercholesterolemia and Dyslipidemia," in Goodman & Gilman's The Parmacological Basis of Therapeutics 12th Ed, pp. 877—906, 2011.
[134] "ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinum," The Journal of Antibiotics, vol. 29, pp. 1346—1348, 1976.
[135] "Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent.," Proceedings of the National Academy of Sciences, vol. 77, pp. 3957—3961, 1980.
[136] "Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials," The Lancet, vol. 376, pp. 1670—1681, 2010.
[137] "Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins," The Lancet, vol. 366, pp. 1267—1278, 2005.
[138] "Statin therapy: review of safety and potential side effects," Acta Cardiologica Sinica, vol. 32, pp. 631, 2016.
[139] "Effects of intense low-density lipoprotein cholesterol reduction in patients with stroke or transient ischemic attack: the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial," Stroke, vol. 38, pp. 3198—3204, 2007.
[140] "The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials," Lancet, vol. 380, pp. 581—590, 2012.
[141] "Cost-effectiveness of lipid-lowering treatments in young adults," Journal of the American College of Cardiology, vol. 78, pp. 1954—1964, 2021.
[142] "Statin therapy for young adults: a long-term investment worth considering," Trends in Cardiovascular Medicine, vol. 30, pp. 48—53, 2020.
[143] "Ezetimibe coadministered with simvastatin in patients with primary hypercholesterolemia," Journal of the American College of Cardiology, vol. 40, pp. 2125—2134, 2002.
[144] "Ezetimibe added to statin therapy after acute coronary syndromes," New England Journal of Medicine, vol. 372, pp. 2387—2397, 2015.
[145] "Adding ezetimibe to statin therapy: latest evidence and clinical implications," Drugs in Context, vol. 7, 2018.
[146] "Ezetimibe therapy: mechanism of action and clinical update," Vascular Health and Risk Management, vol. 8, pp. 415, 2012.
[147] "Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans," Circulation, vol. 135, pp. 352—362, 2017.
[148] "PCSK9 inhibitors: clinical evidence and implementation," Nature Reviews Cardiology, vol. 16, pp. 155—165, 2019.
[149] "Apolipoprotein B, residual cardiovascular risk after acute coronary syndrome, and effects of alirocumab," Circulation, vol. 146, pp. 657—672, 2022.
[150] "PCSK9 inhibitors," Current Opinion in Lipidology, vol. 24, pp. 251—258, 2013.
[151] "Are PCSK9 inhibitors the next breakthrough in the cardiovascular field?" Journal of the American College of Cardiology, vol. 65, pp. 2638—2651, 2015.
[152] "Effect of Evolocumab on Non-High-Density Lipoprotein Cholesterol, Apolipoprotein B, and Lipoprotein (a): A Pooled Analysis of Phase 2 and Phase 3 Studies," Journal of the American Heart Association, vol. 9, pp. e014129, 2020.
[153] "Safety and tolerability of PCSK9 inhibitors: current insights," Clinical Pharmacology: Advances and Applications, vol. 12, pp. 191, 2020.
[154] "PCSK9 Inhibitors and ezetimibe monotherapy in patients not receiving statins: a meta-analysis of randomized trials," Current Vascular Pharmacology, vol. 19, pp. 390—397, 2021.
[155] "How much should LDL cholesterol be lowered in secondary prevention? Clinical efficacy and safety in the era of PCSK9 inhibitors," Progress in Cardiovascular Diseases, vol. 67, pp. 65—74, 2021.
[156] "Cardiovascular efficacy and safety of PCSK9 inhibitors: systematic review and meta-analysis including the ODYSSEY OUTCOMES trial," Canadian Journal of Cardiology, vol. 34, pp. 1600—1605, 2018.
[157] "PCSK9 inhibitors and ezetimibe with or without statin therapy for cardiovascular risk reduction: a systematic review and network meta-analysis," Bmj, vol. 377, 2022.
[158] "Serious adverse events and deaths in PCSK9 inhibitor trials reported on ClinicalTrials. gov: a systematic review," Expert Review of Clinical Pharmacology, vol. 13, pp. 787—796, 2020.
[159] "PCSK9 inhibitors: a technology worth paying for?" U.S. Patent 3, 2016.
[160] "Updating cost-effectiveness—the curious resilience of the $50,000-per-QALY threshold," New England Journal of Medicine, vol. 371, pp. 796—797, 2014.
[161] "The role of fibrates in managing hyperlipidemia: mechanisms of action and clinical efficacy," Current Atherosclerosis Reports, vol. 6, pp. 148—157, 2004.
[162] "Lipid-lowering drugs in the management of hyperlipidaemia," Pharmacology & Therapeutics, vol. 79, pp. 205—230, 1998.
[163] "Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy," New England Journal of Medicine, vol. 365, pp. 2255—2267, 2011.
[164] Goodman & Gilman's The Parmacological Basis of Therapeutics 12th Ed, pp. 0—2084, 2011.
[165] "The association between dietary cholesterol intake and subclinical atherosclerosis in Korean adults: The Kangbuk Samsung Health Study," Journal of Clinical Lipidology, vol. 11, pp. 432—441, 2017.
[166] "Meta-regression analysis of the effects of dietary cholesterol intake on LDL and HDL cholesterol," The American Journal of Clinical Nutrition, vol. 109, pp. 7—16, 2019.
[167] "Increases in dietary cholesterol are associated with modest increases in both LDL and HDL cholesterol in healthy young women," Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 15, pp. 169—178, 1995.
[168] "Reducing saturated fat intake is associated with increased levels of LDL receptors on mononuclear cells in healthy men and women," Journal of Lipid Research, vol. 38, pp. 459—468, 1997.
[169] "Dietary cholesterol and cardiovascular risk: a science advisory from the American Heart Association," Circulation, vol. 141, pp. e39—e53, 2020.
[170] "Exploring the factors that affect blood cholesterol and heart disease risk: is dietary cholesterol as bad for you as history leads us to believe?" Advances in Nutrition, vol. 3, pp. 711—717, 2012.
[171] "Effect of dietary cholesterol on low density lipoprotein-receptor, 3-hydroxy-3-methylglutaryl-CoA reductase, and low density lipoprotein receptor-related protein mRNA expression in healthy humans," Lipids, vol. 33, pp. 1177—1186, 1998.
[172] "Existence of consistent hypo-and hyperresponders to dietary cholesterol in man," American Journal of Epidemiology, vol. 123, pp. 221—234, 1986.
[173] "Polymorphisms in ABCG5/G8 transporters linked to hypercholesterolemia and gallstone disease," Nutrition Reviews, vol. 66, pp. 343—348, 2008.
[174] "Dietary cholesterol and plasma lipoprotein profiles: randomized controlled trials," Current Nutrition Reports, vol. 2, pp. 274—282, 2013.
[175] "Episode 185: Cardiovascular disease & why we should change the way we assess risk," The Drive Podcast, 2021.
[176] "Hematopoiesis," in Basic Histology Text and Atlas, pp. 256, 2016.
[177] "Elements of the immune system and their roles in defense," in The Immune System 4th Ed, pp. 14, 2015.
[178] "Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells," Circulation, vol. 113, pp. 691—700, 2006.
[179] "Rationale for the role of osteoclast-like cells in arterial calcification," The FASEB Journal, vol. 16, pp. 577—582, 2002.
[180] "Multinucleated giant cells in atherosclerotic plaques of human carotid arteries: Identification of osteoclast-like cells and their specific proteins in artery wall," Experimental and Molecular Pathology, vol. 99, pp. 654—662, 2015.
[181] "Carbonic anhydrases II and XII are up-regulated in osteoclast-like cells in advanced human atherosclerotic plaques—Tampere Vascular Study," Annals of Medicine, vol. 42, pp. 360—370, 2010.
[182] "Basic mechanisms in atherosclerosis: the role of calcium," Medicinal Chemistry, vol. 12, pp. 103—113, 2016.
[183] "TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify," The Journal of Clinical Investigation, vol. 93, pp. 2106—2113, 1994.
{kind=link}
{kind=link}